The Important Role that Biopharmaceutics Plays in Accelerating Early-Phase Drug Development

Biopharmaceutics is a scientific discipline that examines the interrelationship of the physicochemical properties of the drug, the dosage form in which the drug is given, and the route of administration on the rate and extent of systemic drug absorption (Applied Biopharmaceutics and Pharmacokinetics, Shargel, Wu-Pong and Yu, 5th Edition).
Quotient Sciences helps biotech and pharma customers in the development and optimization of drug products. Our chemists and formulation scientists review the properties of new drug candidates and “work their magic” to develop formulations that improve the exposure profile of the compound.
Many compounds present sub-optimal pharmacokinetic (PK) data (either predicted from in-vitro and pre-clinical data or measured in the clinic), such as poor exposure (leading to high doses), large variability, short half-life requiring more than once-a-day dosing, or Cmax-related adverse events (AEs). Poor exposure and/or large variability can often be addressed and improved upon with enabled formulations to enhance solubility, such as an amorphous spray-dried dispersion (SDD) formulation or lipid formulations. For compounds with large peak-to-trough ratios, more than once-a-day dosing, or Cmax-related AEs, a modified-release (MR) formulation could often be used to successfully alter the input rate and hence modify the shape of the profile to deliver the required PK exposure profile.
To embark on formulation optimization, be it solubility enhancement or MR development, it is key that we understand the biopharmaceutic properties of the compound to guide the formulation strategy and technology selection.
How does bioavailability play a role in biopharmaceutics?
As formulators, we want to deliver the right amount of drug at the right time with the correct concentration within the body to exert a therapeutic effect. We need to understand systemic exposure of the drug, and for an orally administered formulation, that means understanding the process of absorption and then teasing apart the rate-limiting steps in the process.
Biopharmaceutics allows you to understand the solubility, dissolution, and permeability of a compound, and from this we can then assess the potential fraction absorbed (Fabs). Now fraction absorbed and bioavailability are often confused and used interchangeably. Fraction absorbed is directly related to the solubility, dissolution, and permeability of a compound and is the amount of drug that enters the intestinal enterocyte in our gastrointestinal tract (FDA definition), whereas bioavailability (F) is the amount of drug in the systemic circulation able to have a therapeutic effect. F is directly related to the amount of drug absorbed (Fabs) and the amount surviving first-pass metabolism. Therefore, absorption is the input mechanism and clearance (metabolism) is the output mechanism. As formulators, we are often able to directly impact the amount of drug absorbed through formulation optimization and improve exposure. However, the chances of improving the exposure profile of a drug that is highly cleared by formulation modification are limited.
How can biopharmaceutics help drug developers overcome challenges with their small molecules?
Understanding the biopharmaceutic properties of your compound can help you identify a formulation strategy that overcomes the challenges the compound faces or can assess the potential for the specific compound to meet the target product profile (TPP). The sooner challenging and unfixable compounds are identified and killed in development, the less R&D expenditure will be incurred, allowing you to focus on compounds that have the legs to make to it market.
For example, if drug X has a low Fabs of 10% and F is 8%, then there is the option to increase Fabs through formulation optimization. However, if drug Y has a high Fabs (90%) but low F (e.g. 10%), even if we are able to increase absorption by another 10% (Fabs = 100%), it is unlikely to improve the exposure (F) greatly, as for drug Y clearance (metabolism) is limiting exposure. The only instances in which formulators can help in this scenario is to increase exposure (Fabs) through formulation just enough to potentially saturate the clearance mechanism. Alternatively, if the compound is subject to gut CYP3A4 metabolism, we could deliver to a lower region of the gastrointestinal tract where CYP3A4 expression is reduced, thus hoping to bypass the gut metabolism if that is the rate-limiting process for exposure. However, often in this situation it is back to discovery and the drawing board to revisit the compound chemistry.
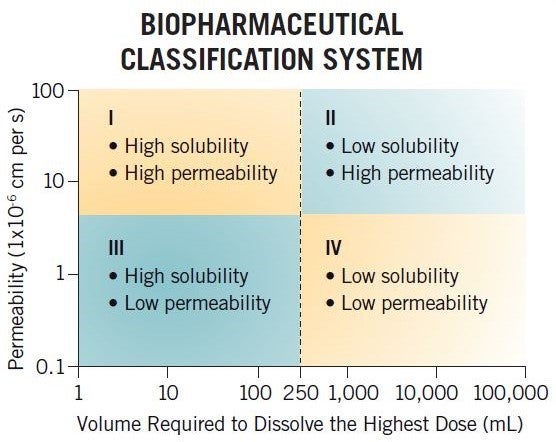
What is the Biopharmaceutics Classification System (BCS)?
The BCS is a regulatory tool that is used to justify clinical biowaivers for certain types of compounds (BCS Class I and III) based on dissolution data, allowing sponsors to justify not performing clinical bioequivalence studies when changing a formulation. The framework classifies compounds based on their permeability and solubility (buffer solubility) properties into four categories (BCS I, II, III, and IV), and this system has been used by the industry for many years to assess in-vivo performance.
For example, a BCS Class I compound with high solubility and high permeability is likely to be a good development candidate due to having high fraction absorbed. However, a BCS Class IV compound is not thought of in such good light, having low permeability and low solubility and hence thought to have poor exposure. In reality, a BCS Class IV compound could have Fabs of 80% and high solubility at pH 6.5, and therefore have good Fabs and no formulation development issues.
The BCS classification criteria are strict and hence often misinform clients of their compound’s formulation/development challenges. More recently, a classification system based on developability potential has been developed by Dressman and Butler, the Developability Classification System (DCS). This classifies compounds into four categories similar to the BCS, but uses simulated intestinal media for the solubility assessment and also takes into consideration the compensatory nature of permeability, allowing a solubility-limited absorbable dose to be determined, which in turn allows for DCS II compounds to be divided into DCS IIa and DCS IIb compounds. DCS IIa compounds are dissolution limited and hence formulation strategies to improve exposure would focus on particle size reduction such as nanomillling and micronization, whereas DCS IIb compounds are solubility limited and hence solubility-enhancement strategies such as SDDs and lipids may be used to improve exposure.
How can the DCS be used to drive formulation strategies?
Quotient Sciences uses the DCS to help drive formulation strategies for our clients. We can either take existing customer data and assign a DCS classification or measure solubility and calculate a predicted human effective permeability (Peff) using GastroPlus® ADMET predictor, which is done by the modeling and simulation group based on the compound structure.
A recent example of this was for a compound at the candidate selection stage. Quotient Sciences supported a standalone DCS classification and formulation development package. Permeability was high and solubility at 24 hours in intestinal buffer was less than the expected therapeutic dose, so solubility was classified as “low”. However, solubility at 3 hours was found to be >10-fold higher and hence it was classified as a DCS IIa compound. So, if dissolution is rapid, absorption will be good and sophisticated solubility-enhancement strategies are not required. Quotient Sciences then developed a simple capsule formulation with particle size reduction (micronization) and wetting agents to support the first-in-human (FIH) clinical study.

How can biopharmaceutics be leveraged as part of an integrated development program?
Drug developers have also seen many benefits in utilizing an integrated approach in early-phase drug development in order to minimize downstream risks, reduce R&D spend, and accelerate time to clinic. Quotient Sciences’ unique Translational Pharmaceutics® platform integrates drug substance, clinical manufacturing, and clinical testing activities under a single organization, and allows formulation optimization within a clinical study, rather than the traditional approach of pre-clinical testing to select a lead formulation. We all know that the correlation between pre-clinical exposure and exposure in man for many compounds is questionable1, hence if formulation selection is performed in rat or dog, you run the risk of it not performing in man.
Quotient Sciences uses the biopharmaceutics information along with the DCS to select a formulation strategy, and then within a study will make and test small batches (approximately 200 units) of drug products with limited stability (usually only 7 days) in the clinic. Essentially, we are doing our formulation selection or optimization in man rather than rat and dog. This approach saves considerable time and money, as demonstrated in the recent article by the Tufts Center for the Study of Drug Development (CSDD).
The benefit of this approach is highlighted by a recent poster publication with Boston Pharmaceuticals, where Quotient Sciences developed three formulations for a problem compound. In a previous FIH Phase I study, BOS172767 had shown poor and variable exposure with a large food effect, which had resulted in clinical progression being halted. Quotient Sciences developed a micronized capsule, a lipid capsule, and an SDD tablet for BOS172767, which were tested in the clinic, and all showed improved exposure compared to the reference capsule formulation. The SDD tablet had the highest exposure, but the micronized capsule was selected for progression, as Cmax was lower (which could be advantageous for Cmax-related side effects), AUC was similar, and commercial progression of a micronized capsule formulation is easier and cheaper than an SDD formulation. The micronized capsule was progressed to Part 2 of the study and showed a linear increase in exposure (AUC) up to 800 mg, establishing safety margins for future clinical testing.
The Translational Pharmaceutics approach of testing multiple formulations within a clinical trial can also be applied to FIH studies. This results in sponsors exiting FIH with not only safety, tolerability, and PK data, but also having performed formulation selection. In the case of Rigel Pharmaceuticals’ R552, two SDD suspension formulations were assessed during a single-ascending-dose (SAD) study. The lead SDD suspension was then progressed to the remaining SAD cohorts and multiple-ascending-dose (MAD) study, but at the same time, Quotient Sciences developed an SDD tablet in parallel. In Part 3 of the study, which investigated relative bioavailability and food effect, the SDD tablet was assessed in the fed and fasted state and compared back to the reference SDD suspension. This meant that at the end of FIH, Rigel had a suitable solid oral dosage form for Phase II patient studies, which Quotient Sciences then supplied to the relevant clinics, reducing time to proof of concept (POC). Combining biopharmaceutics, DCS assessment, and Translational Pharmaceutics allowed successful formulation selection and optimization within a clinical trial.
Summary
In summary, biopharmaceutics underpins the formulation strategies used at Quotient Sciences, ensuring a science-based and data-driven approach to formulation optimization. This reduces the risk of drugs failing due to poor formulation and increases the chances of clinical success.
Watch our on-demand webinar, “Getting It Right Early – Overcoming Challenges for Small Molecules”, for more information on how biopharmaceutics can help accelerate your early-phase drug development program, or connect with us today to discuss your program.
References
- G. M. Grass and P. J. Sinko, “Effect of diverse datasets on the predictive capability of ADME models in drug discovery” Drug Discovery Today 6(12) (Suppl.) S54 – S61 (2001).
Source link
#Important #Role #Biopharmaceutics #Plays #Accelerating #EarlyPhase #Drug #Development