Recently, mRNA technology has emerged as a powerful therapeutic and drug delivery modality. It has the potential to replace proteins, induce stem cells and even facilitate genome engineering.
The Moderna and Pfizer/BioNTech COVID-19 vaccines served as compelling evidence of the potential for mRNA-LNP therapeutics on a global scale, demonstrating that lipid nanoparticles (LNPs) can be formulated to protect mRNA, aid transport into target cells in the body and release therapeutic mRNA into the cytoplasm. In some cases, LNPs also have a therapeutic effect, serving as adjuvants and enabling additional immunostimulation.
Meanwhile, mRNA-based candidates for cancer immunotherapies, autoimmune disorders and other infectious disease vaccines have entered clinical development, some with promising initial results. In addition, LNPs are being explored as a delivery system for CRISPR/Cas9 gene therapies in human clinical trials.
GlobalData’s Pharmaceutical Intelligence Centre database shows there are an estimated 1,295 RNA-based drugs in development worldwide, racing to transform the treatment landscape for many different types of patients and diseases. Currently, almost 1,700 clinical trials are in progress, exploring the efficacy of RNA-based drugs for therapeutic areas ranging from oncology to the central nervous system, cardiovascular, musculoskeletal, and infectious diseases such as HIV and Hepatitis.
This has turned attention to the delivery systems required to transport mRNA to the cell site safely and efficiently. As a result, LNP formulations as a delivery system are gaining traction.
Emergence of LNPs
LNP drug-delivery formulations are geared to support delivery of a targeted drug substance and can be fine-tuned to optimise delivery, and thus therapeutic effect. Due to the environmental pH sensitivity of ionizable lipids, LNPs function as an effective delivery system for nucleic acids by protecting the RNA cargo from degradation and mediating safe endogenous cellular uptake. Many scientists believe RNA-LNP technology is the future of medicine, offering improved and even game-changing outcomes for patients.
GlobalData analysis shows 152 RNA-LNP drugs are in development, providing important new avenues for developing therapeutics for serious and difficult to treat conditions, such as infectious diseases and cancers.
Figure 1: RNA-LNP drugs in development
LNPs have the potential to deliver a variety of existing and novel therapeutics that are important to biotech and established biopharma, and can be used in the emerging field of gene-editing technology.

Industry players wanting a piece of this action, however, are faced with the daunting challenge of navigating a rapidly evolving regulatory framework. While an optimised regulatory strategy can result in much faster and less complex time to market, any missteps can endanger a product’s development.
One of the greatest strengths of LNP formulations – and the largest hurdle to overcome – is the ability to customise and fine-tune each one to optimise delivery for a specific therapeutic drug. They are not one-size-fits all, so each developer has the opportunity to optimise the LNP for their specific application. However, they may also face unique manufacturing challenges and regulatory pitfalls as they progress from process development to a clinical good manufacturing practice (GMP) product that can be used in humans.
For example, an LNP formulation optimised for infectious disease vaccine applications may not be optimal for cell therapies, and vice versa. These LNPs are made up of four key components, including the ionizable lipid that complexes the therapeutic drug (mRNA, for example), the stabiliser lipid, cholesterol, and helper phospholipid that assists with preventing aggregation. Alterations in the chemical characteristics of these components, or their ratios with each other, will affect the efficiency, performance and biodistribution of the therapeutic drug being delivered.
In a recent study by Precision NanoSystems, a LNP innovator and solutions provider, scientists evaluated the immunogenicity of saRNA-LNP COVID-19 vaccine candidates in non-naive non-human primate (NHP) models (rhesus monkey). The World Health Organisation SARS-CoV-2 human IgG standard was used to establish a quantitative curve for relative quantification of anti-spike IgG in serum.
“In non-naive rhesus monkey NHP models, the saRNA-LNP vaccine demonstrated an immune response, high IgG levels, good safety, and did not appear to be genotoxic while testing the immunogenicity of the saRNA Covid vaccine,” says Dr Anitha Thomas, Director of R&D at Precision NanoSystems.
This is just one of many optimised ionizable lipids available in Precision NanoSystems’ LNP portfolio that support a wide array of therapeutic applications that are in development by academic and commercial partners, saving them significant time in candidate selection, product optimisation and the choice of process parameters.
Navigating regulatory challenges for LNPs
A central challenge in LNP drug-delivery formulations is navigating their various global considerations in regulatory filings, both in the regulatory review of clinical trials and regulatory approval processes. Drug developers must navigate a complex framework of guidance and requirements specific to their therapeutic class and indication, as well as those pertaining to LNPs specifically. This requires careful integration of regulatory strategy into the development process.
For oligonucleotide drug regulatory development and approvals, there remain questions of how “sameness” of active pharmaceutical ingredients (APIs) will be defined, and there is currently no consensus US, EU, or International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidance for determining if an oligonucleotide API will be considered the same in terms of its biochemical properties and manufacturing process for the purposes of approval of future generic drugs. In 2022, the European Medicines Agency (EMA) released a concept paper outlining the need for more detailed guidance on development and manufacture of oligonucleotide drugs. Following the 2022 release of its guidance on clinical pharmacology in oligonucleotide therapeutics, speakers from the United States Food and Drug Administration (FDA) also highlighted that additional guidance and harmonised regulatory standards will be important in advancing future novel oligonucleotide formulations and enabling regulatory approvals of generic oligonucleotide drugs in this space. With multiple regulatory agencies highlighting a need for additional guidance, it is very likely that new guidance affecting oligonucleotide therapeutic development using LNPs will be developed in the foreseeable future.
As well as navigating regulatory approvals, vaccine and therapeutic developers using a range of formulations with LNP components must navigate regulatory submissions required early in development before they can begin clinical testing of these novel investigational products in humans. This is done through a regulatory review process called Investigational New Drug (IND) applications in the US or Clinical Trial Applications (CTA) in most global regions. These regulatory filings are also required to test new formulations, indications or dosages of existing drugs in humans. Regulatory filings can present a significant time and resource burden for pharmaceutical developers.
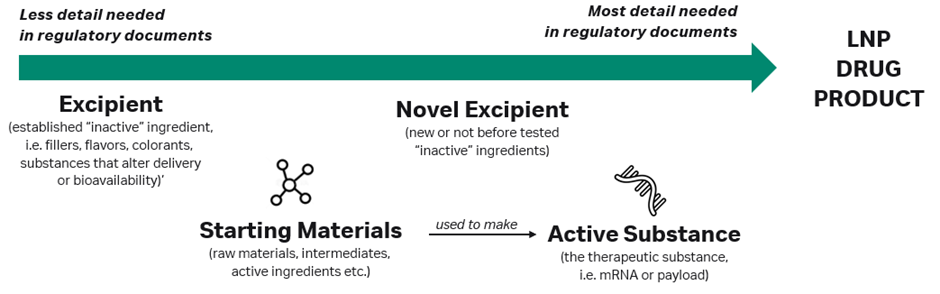
Both regulatory approval and clinical trial submissions follow strict content and formatting requirements, which require classifying each component of a new drug based on its use in the product. The therapeutic modality (i.e. mRNA) is considered to be an active substance (or an API) because it exerts a therapeutic effect on the body. Starting materials are the various raw materials and intermediates used to make an API. Many other inert substances in drug formulations are considered excipients, such as colours, flavourants, fillers and substances that improve drug delivery but do not otherwise have their own therapeutic effect on the body.
For the majority of LNPs used in drug delivery, regulatory agencies may consider the LNP component to be a ‘novel excipient’, although in certain cases (such as immunomodulating) the LNP may be treated as an active substance. In other cases, particularly where innovative new lipid formulations are used, regulators may treat each lipid in the formulation as a ‘unique established’ or ‘novel’ excipient.
Global regulatory agencies do not always agree on their approach, resulting in novel LNP products sometimes being treated as active substances in one region and as novel excipients in another, further complicating the process of preparing regulatory applications to start clinical trials in humans.
Generally, established excipients are those already used in previous products and recognised by a regulatory agency, meaning that developers of new LNP products only need to include minimal information about these in regulatory filings since their purpose and safety are well known already. Novel excipients will require extensive documentation on manufacturing, characterisation, purity profile and more to satisfy the regulatory body that it is well-characterised. Active substances require even more comprehensive documentation that explores their toxicity and mechanism of action (see Figure 3).
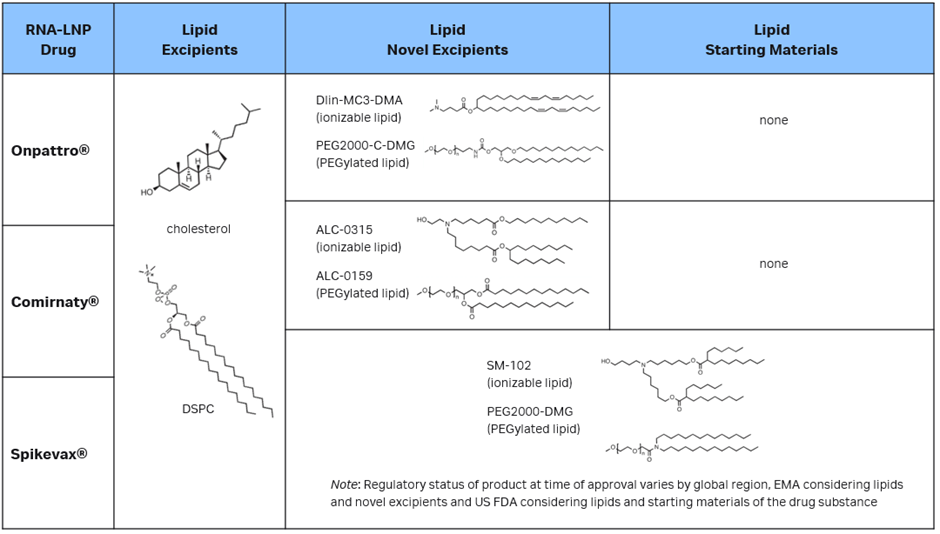
This is seen in commercial LNP drug products in the debate between active substances and excipients. We need only look at the three approved LNP COVID-19 vaccine products: Onpattro®, Spikevax® and Comirnaty®. All three are approved for use in humans by regulators, including the FDA and the EMA. Despite their very similar LNP formulations, Spikevax’s LNP components are classified differently than the cognate LNPs in the other two products.
In its initial submission for Spikevax, Moderna considered both the lipid components and mRNA to be active substances. But, at the request of the EMA, the regulatory filing was modified to consider only mRNA to be an active substance and all four lipid components as excipients (two of which are considered novel: the SM-102 ionizable lipid excipient and the polyethylene glycol-lipid conjugate PEG2000-DMG). Conversely, the US FDA considered PEG2000-DMG and SM-102 as “starting materials” for the drug substance and not excipients. In its FDA approval, the Chemistry Manufacturing and Controls (CMC) BLA Review Memorandum explicitly states that the product does not contain any novel excipients.
In both the US and Europe, the Onpattro and Comirnaty LNP vaccines contain four pharmaceutically inactive lipid excipients (two of which are considered novel excipients) in addition to their respective active substances. In brief, these regulatory actions establish the precedent that, based on the formulation, a regulator may consider lipids in LNP formulations as excipients or drug substances, whereas very similar lipids in Onpattro and Comirnaty were reviewed as excipients. The EMA was more consistent in its review, as the lipids in all three LNPs are listed as excipients, novel excipients, starting materials for an active substance, or potentially even active substances in their own right if exerting a biopharmaceutical effect.
Note: details discussed on chemical structure and lipid classification within various commercial LNP products can be found in the European Public Assessment Report and US FDA publicly accessible review and approval documents at Drugs@FDA (FDA approval letter, labels, summary basis for regulatory action).
These three RNA-LNP therapeutics have set the precedent that lipid components can be treated as either established or novel excipients, but in some cases other classifications – and, correspondingly, more information and testing in the regulatory documentation – may be requested by regulatory agencies either before a new LNP drug product can be tested in human clinical trials or before its approval.
One strategy to streamline both the clinical trial (IND/CTA) regulatory process and the ultimate drug approval is to establish the level of information needed by regulators for each component of the formulation early on in preclinical development. In the US, this can be done at meetings such as the Pre-IND several months before filing the IND, but it is often a better strategy to work closely with your contract development and manufacturing organisation (CDMO) much earlier, through Initial Targeted Engagement for Regulatory Advice on CBER Products (INTERACT) meetings or the new Type D meetings, which allow for early conversations about manufacturing and process development, and the requirements for future regulatory documents. In Europe, both the EMA and country-level competent authorities have similar scientific advice meetings available in the months before filing a CTA, and even earlier innovation meetings.
By leveraging early partnerships between formulation and process developers, CDMOs and early legal interactions, industry developers can de-risk and streamline future LNP drug product regulatory milestones.
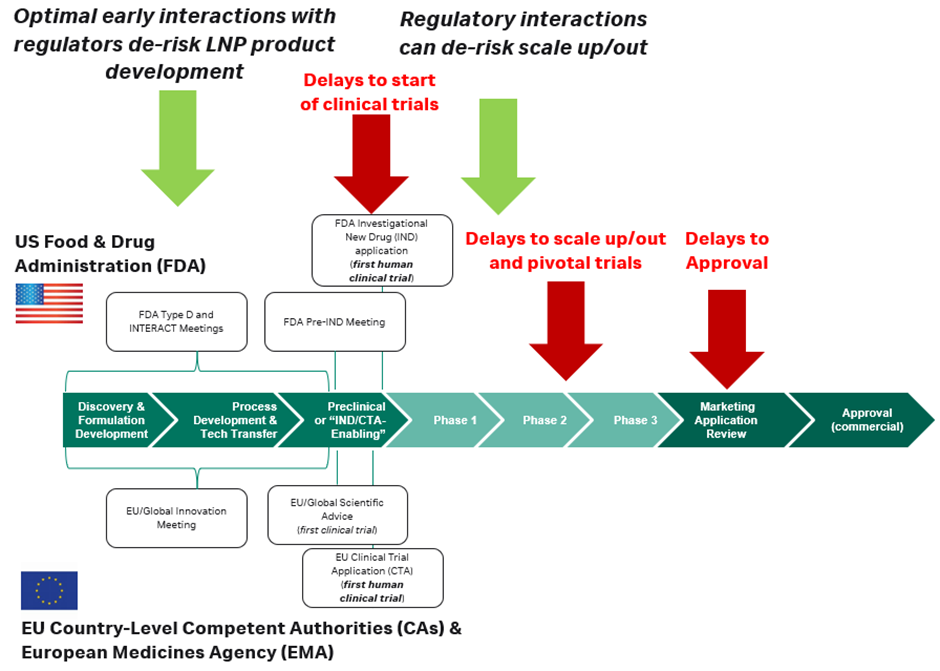
Developer interactions with regulatory authorities after clinical trials have been completed at each phase, or ‘end-of-phase’ meetings, and conducted ad hoc can also de-risk changes in process development and scale up/out throughout development. Lipid nanoparticles are surface active, designed to interact with membranes with flexibility and mobility. When manufactured in large volumes or under GMP conditions, they can be affected by filtration processes or aseptic filling.
While it might not be needed to start early-phase or first-in-human trials, as the product progresses into the later-phase trials and approval, regulators will require analytical data on the RNA and LNP condition, often in the form of comparability studies, which can add significant time and complexity to development. This is carried out to show that from early development to final approval, formulations do not significantly change the key attributes of the drug, such as its potency. It is recommended to have an internal expert monitor the analytics or partner with experts who can provide guidance for scaling formulations and processes.
RNA-LNPs may also be eligible for special regulatory pathways, such as Orphan Drug or Rare Disease status, Accelerated Approval, Breakthrough Therapy Designation, Fast Track, Advanced Therapy Medicinal Product designation, and other special programmes open to certain types of products and developers. The benefits of these programmes include rolling reviews, more frequent interactions with regulators, and other development incentives such as competitive protection through exclusivity and tax benefits.
As more LNP and novel products that share underlying scientific characteristics are developed, regulators are developing new approaches to help expedite ‘platform’ technologies that share common scientific elements. One commonly used strategy among developers with multiple pipeline candidates sharing a similar technology is to cross-reference IND/CTA or approval dossiers, so complex documents reviewed for a prior product can be reused by the developer to streamline preparation and review.
More recently, as part of the 2023 Omnibus Appropriations Act in the US, the FDA established a new formal regulatory process called the Designation Program for Platform Technologies. The PREVENT Pandemics Act includes a similar designation programme for platform technologies that have the potential to increase efficiencies in drug development. These new programmes help to harmonise requirements and streamline the regulatory process for novel platforms, and they will be important strategic aspects of future LNP product development.
While regulators gain a better understanding of LNP products, it remains that the route of administration, dosing regimen and application affect not only formulations but also how LNPs are classified in regulatory documents and the complexity of regulatory filings. It is important to understand the current landscape and strategically interact with regulators early, and throughout development, to ensure scientific data is collected to support any novel therapeutics or modalities.
Paving the way forward
When developing a new therapeutic product using non-viral delivery with LNPs, formulations can be optimised through in-house development or outsourcing. Typically, building an internal team and capabilities takes years, so acquiring the technology (ionizable lipids, formulations and services) through a partner is a great way to speed up programmes.
It is critical to recognise that in RNA-LNP development – as with other innovative products and platforms – each formulation and its optimal regulatory strategy will be unique. While most developers have legal and clinical research partners later in development, establishing an early regulatory strategy, and partnering LNP manufacturing and legal experts to interact with regulators, can streamline development. This enables faster first-in-human trials and reduces the risk of approval being delayed. Such partnerships can help developers stay abreast of the changing regulatory landscape. Ultimately, early and timely communications with regulators, and partnering with experts to navigate product and regulatory processes, is essential to enable developers to bring safe therapies to patients faster.
Vancouver-based Precision NanoSystems is a leading provider of nanoparticle technologies, reagents, services and strategies for the development of genomic medicines. In July 2022, its ionizable lipid portfolio and scalable microfluidic manufacturing platform were endorsed by life sciences startup Replicate Bioscience, a company pioneering novel self-replicating RNA (srRNA) technology for use with infectious diseases, oncology and autoimmune diseases.
Under a licensing agreement with Replicate, Precision NanoSystems will provide LNP solutions for the scale-up and manufacture of 15 Replicate srRNA therapeutics. When announcing the agreement, Andy Geall, co-founder and chief development officer of Replicate, said Precision NanoSystems would enable Replicate to expand its portfolio of srRNA therapeutics beyond its lead candidates nominated for human trials.
The LNP landscape is dynamic. As more therapeutics for various applications enter clinical trials, awareness will increase in the RNA-LNP field and push technology into new areas, which will create new opportunities for development.
“One of our unique offerings is our GenVoy-ILMTM delivery platform, through which developers can access a well-characterised LNP portfolio, comprising ready-to-use LNP reagent kits and an ionizable lipid library, designed to accelerate genetic medicine development,” says Dr Lloyd Jeffs, Senior Director of Pharmaceutical Development.
“Access to quality lipids and optimisation expertise can provide a competitive advantage, fast-tracking development by reducing the number of lipid formulation steps, simplifying the process and reducing cost.”
“Our BioPharma Services team supports developers to optimise compositions for testing in a specific biological model, and our NanoAssemblr® microfluidic mixing technology allows for nanoparticles to be produced with the same critical quality attributes across scales, from early preclinical to commercial manufacturing. In addition, our world-class regulatory solutions teams can help you strategically roadmap your milestones and facilitate regulatory interactions, leveraging our experience from decades of successful global experience.”
He adds: “Once lead candidates are selected, we work collaboratively to iterate the LNP formulations and scale-up process, with supporting analytics and CMC guidance. Given the fast-evolving landscape of RNA-based medicine and the infancy of the use of LNPs as a delivery system, it really is imperative for developers to work with partners such as us to speed up time to market.”
Source link
#RNALNPs #Navigating #regulatory #challenges